
Understanding how apoE contributes to the clearance and metabolism of Aβ in Alzheimer’s disease
The Paul Laboratory studies the fundamental disease biology of Alzheimer’s disease with the ultimate goal of translating this understanding to the discovery and development of disease-modifying therapies. The work in our laboratory is multifaceted and involves very basic cellular-molecular studies using a variety of cell-based in vitro models (both of neurons and glia) as well as in vivo work involving the creation and characterization of a number of transgenic mouse models of Alzheimer’s disease-related neuropathology.
Our approach is to begin by gaining a much better understanding of the underlying etiological underpinnings of the disease, namely how do the genes that cause rare autosomal dominant forms of the disease or are major risk factors for late-onset disease (LOAD) actually work? How do these genes / genetic risk factors contribute to the actual pathogenesis of the disease , i.e. how do they lead to the neuropathological hallmarks (amyloid plaques, neurofibrillary tangles, brain inflammation) which ultimately lead to the neurodegeneration (loss of synapses and neurons) that result in the signs/symptoms of disease (the dementia and other neuropsychiatric manifestations of the disease)? Our work to date has primarily focused on the important LOAD risk factor alleles for apoE. The apoE4 allele is now well recognized as the most important known genetic risk factor for LOAD, with heterozygote carriers and homozygote carriers having a 3-14 fold increased risk compared to apoE3 homozygotes, respectively. Moreover the apoE2 allele is a protective allele, reducing overall risk of developing LOAD by approximately 50%. ApoE4 carriers develop AD earlier the non-apoE4 carriers (approx. 5 years for each apoE4 allele).
Using a number of AD transgenic mouse models, our laboratory, both independently and collaboratively, has shown that apoE is a powerful determinant of brain amyloid burden that occurs during aging, with the apoE4 allele dramatically increasing age-dependent brain amyloid burden, compared to the apoE3 and apoE2 alleles. Using in vivo microdialysis we have shown that apoE primarily influences the “clearance” of amyloid-β peptides (Aβ) from the brain interstitial space/fluid. The reduced brain clearance of Aβ in apoE4-expressing mice is believed to account for the robust effect of this risk factor gene to increase brain amyloid burden during aging in humans. Studies are now underway to better understand how apoE contributes to the clearance and metabolism of Aβ and we have found that both astrocytes and microglia (which express apoE) are able to metabolize Aβ and that the apoE isoforms differentially alter the ability of both astrocytes and microglia to metabolize Aβ (apoE4<<apoE3<apoE2). It appears that apoE4-expressing astrocytes and microglia are less efficient at metabolizing Aβ than those that express apoE3 or apoE2. In related studies we have reported that direct intracerebral administration of the three apoE isoforms via viral-mediated gene delivery to the brains of several amyloid-depositing transgenic mouse models results in similar apoE isoform-dependent effects on brain Aβ/amyloid burden, with the apoE4 gene/protein increasing amyloid burden and the apoE2 gene/protein reducing brain Aβ burden. The latter observation has led to an actual gene therapy strategy and project where using AAV viral vectors we are attempting to deliver apoE2 to various brain regions via direct intracerebral administration as well as via relatively noninvasive intrathecal routes of administration. Our goal is to see whether we can deliver sufficient amounts of apoE2 broadly to the brain to effectively reduce brain amyloid burden. Similar work being carried out collaboratively with the Crystal laboratory is underway in nonhuman primates to determine whether sufficient levels of apoE2 can be achieved in the brain of a much larger species and whether this can be achieved safely (without overt toxicity).
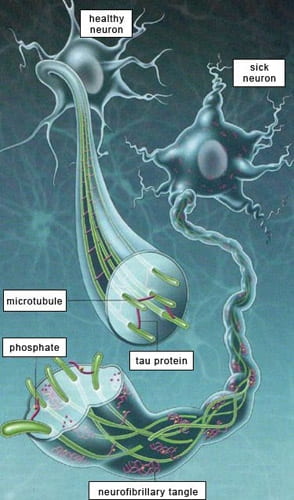
In parallel to these primarily in vivo studies we are also studying the ability of microglia to reduce AD-related neuropathology, including amyloid plaques and neurofibrillary tangles. We have previously reported that microglia (and macrophages) readily degrade both soluble and insoluble Aβ species and do so in an isoform-dependent manner (apoE2>apoE3 >apoE4). More recently, we have discovered that microglia readily phagocytose and degrade various soluble and insoluble tau species. This work coupled with the recently appreciated role of innate immunity in AD risk strongly suggest to us that microglial senescence and dysfunction may account for both AD-related amyloid and tau pathology and may comprise the major etiologically relevant cell type in this disease and possibly other neurodegenerative disorders of aging. Our work is now encompassing “chemical” screening to discover both small and large molecule modulators of microglial function, using in vitro screens established in our laboratory.
Finally, we currently have two collaborative research projects funded through Sponsored Research Agreements with Johnson and Johnson Pharmaceuticals as well as AstraZeneca, both focused on drug discovery related to finding potential disease-modifying therapies for Alzheimer’s disease. Together with our pharmaceutical partners we are making good progress in these efforts and hope to have at least one novel drug candidate progressing to early clinical development by the end of 2015.
In addition to our work on Alzheimer’s disease we are also pursuing a large collaborative NIH-funded study with Dr. Maja Bucan in the Department of Genetics at the University of Pennsylvania on the genetics of bipolar affective disorder. This study, which was first initiated back in 1988 while Dr. Paul was at the NIMH, is attempting to discover genetic risk factors for bipolar affective disorder which is a very serious cyclical mood disorder characterized by episodes of depression (often severe) that are interspersed with periods of mania or hypomania (elevated mood that can also take on psychotic features). Bipolar affective disorder is a highly heritable mood disorder (estimated heritability of 85%) that has very high morbidity and mortality associated with it (very high lifetime risk of suicide in bipolar patients). We have been studying a large multi-generational family with over 450 affected and unaffected members that segregates bipolar disorder from a genetic isolate (Old Order Amish of Lancaster County, Pennsylvania). Family members have been studied longitudinally for many years by Dr. Janice Egeland and her research team and blindly diagnosed by an independent psychiatric review board. Our work over the last 5 years has involved very detailed and thorough genotyping with microsatellite DNA markets, SNPs (microarrays) and whole genome sequencing (60+ subjects sequenced with imputed sequences for the rest of the pedigree). Analyses of this genotypic data have revealed several disease loci and a list of potential candidate genes. These disease gene harboring chromosomal regions and associated genetic variants are being further tested in other Amish and non-Amish pedigrees segregating bipolar disorder in the hopes of discovering one or more genetic variants that account for the high heritability of this disorder.
Work in the Paul laboratory has been generously supported by the Appel Alzheimer’s Disease Research Institute, the National Institutes of Health, The Alzheimer’s Drug Discovery Foundation (ADDF) and through sponsored research agreements with both Johnson and Johnson and AstraZeneca.