
Relationship between synaptic activity and amyloid-beta metabolism in Alzheimer’s disease
Amyloid-beta (Abeta) peptide accumulation within the brain extracellular space, as toxic oligomers and plaques, is strongly believed to be the primary cause of Alzheimer’s disease (AD). My lab focuses on understanding the metabolism Abeta within the brain extracellular fluid, or interstitial fluid (ISF). We developed a novel in vivo microdialysis technique that enables us to specifically measure ISF Abeta within the brains of living and awake wildtype and APP transgenic mice. The technique permits hourly sampling of Abeta for several days, thus providing kinetic information about how Aβ levels change over time under various settings such as aging, behavior, drug treatment, and genetic manipulation. We recently discovered that synaptic activity is a critical regulator of Abeta production in the living brain; as synaptic activity increases ISF Abeta levels rapidly increase and vice versa as synaptic activity declines. This occurs following pharmacologic manipulation of synaptic activity as well as during physiological fluctuations in activity such as sleep/wake cycles or stress. Synaptic transmission causes more clathrin-mediated endocytosis within the presynaptic terminal as synaptic vesicle membrane recycles. As this occurs, APP is internalized into endosomes where Abeta is produced followed by secretion into the ISF. APP is found within the axonal compartment as well as the dendritic compartment. Postsynaptic signaling mechanisms, through a wide variety of receptors including NMDA, muscarinic acetylcholine, and serotonin receptors can modulate APP processing and alter Abeta levels as well.
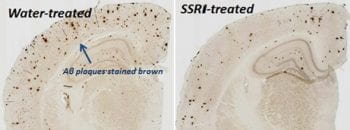
One aspect of our current research is focused on postsynaptic signaling mechanisms that decrease Abeta generation including identification of effectors on the cell surface and the signaling pathways within the neuron that impact Abeta production. Activation of the MAPK/ERK signaling pathway decreases Abeta levels in vivo. ERK can be activated by a multitude of extracellular ligands, including NMDA receptors and serotonin receptors. We can demonstrate the agents that increase serotonin signaling, such as SSRI antidepressants, decrease Aβ levels in the mouse brain which entirely depends on ERK signaling. Inhibiting ERK or MEK (another step in the ERK pathway) completely blocks the SSRI-dependent decrease in Abeta. Work by a collaborator at Washington University, Dr. Yvette Sheline, suggests that SSRI antidepressants reduce plaque load in humans as well. Our hope is that understanding the pathways that control Abeta levels will help us understand risk factors of AD as well as provide new therapeutic targets.
My lab is also developing several new techniques, including in vivo synaptic plasticity assays to determine how various forms of Abeta impact synaptic transmission in a physiological setting. We are also developing new protein-specific electrochemical technologies to measure Abeta in the scale of seconds or milliseconds as opposed to hours currently available with our microdialysis technique. Such an approach would allow us to measure Abeta release from neurons in real-time and further define the complex relationship between synaptic activity and Abeta generation.